由于细胞是生物学的基本单位,研究人员正更加努力地尝试将它们进行单个分离、研究和比较。DNA研究主要涉及的是测序单细胞微生物相对简单的基因组;而更大更复杂的人类细胞基因组则是一个更大的挑战。随着测序成本的大幅度下降,破译来自单细胞的30亿碱基的基因组并逐个细胞比较序列正在变为现实。
在过去的几十年里,研究人员对于来自许多物种的成千上万的基因进行了测序。几乎在所有情况下,破译的DNA都是从数百万的细胞中提取出来并混合在一起。其结果是大量的数据,但是博德研究所的遗传学家Joel Hirschhorn说:“当你看向的是整个细胞群时,有许多的现象其中包括大量细胞与细胞间的变异就会变得不再明显。”这有可能会改变。
在会上,四个小组描述了关于基因组如何生成多样性和什么导致特异肿瘤耐受治疗的新见解——所有的都是来自单个人类细胞DNA测序的结果。他们的报告引发了科学家对单细胞测序潜力的兴奋。“我们才刚刚开始触及表面。”Hirschhorn说。
由于细胞是生物学的基本单位,研究人员正更加努力地尝试将它们进行单个分离、研究和比较(Science, 7 January 2011, p.24)。DNA研究主要涉及的是测序单细胞微生物相对简单的基因组;而更大更复杂的人类细胞基因组则是一个更大的挑战。随着测序成本的大幅度下降,破译来自单细胞的30亿碱基的基因组并逐个细胞比较序列正在变为现实。
对于艾伯特·爱因斯坦医学院的统计遗传学家Adam Auton而言,单细胞测序提供了一个了解重组(细胞分裂过程中配对染色体交换DNA片段的过程)的窗口。重组通过将各种版本的基因一起放置在新组合中帮助生成了遗传多样性。它也是细胞剔除异常DNA的一种方法。
研究人员长期以来一直在寻求一种途径来测定发生在人类重组量,他们提出了一些间接的方式在家庭中或在群体中对其进行测量。“测序和比较单个精子细胞的基因使得直接且更为系统地探究了这一现象。”Auton说。
通过对单个精子细胞测序,研究人员正在解析重组率
因此,当中国强大的测序机构北京基因组研究所(Beijing Genomics Insititute)告诉Auton已经完成了对一名50岁年纪亚洲男性的167个精子的测序时,Auton热切地想要看到数据。他初期的分析突出了分离单细胞以及可靠地拷贝DNA进行测序是多么的困难。研究人员发现了若干来自多种细胞的DNA序列;然而只有40个达到了标准。通过将每个精子的DNA与来自血液样本的DNA进行比较,Auton和他的同事们检测到了近1000次重组事件,每个精子细胞约24.5次,这一数字与来自间接方法的预测相一致。重组在整个染色体不均衡发生,一些“热点”(hot spot)代表了重组率增高位点。大多数情况下,Auton发现的热点与研究人员在其他研究中看到的相一致。然而在15号染色体上也有一个重组波峰,Auton在会议上报告说。
来自斯坦福大学的Stephen Quake一直在利用一种所谓的芯片实验室(lab on a chip)开发单细胞技术。样本通过一连串微通道和微阀门分离单个细胞提取出DNA,并拷贝数千次以获得足够的量用于测序。Quake报告说他已经解决了许多技术问题。他在去年已经完成了人类的单细胞测序,现在他正利用这项技术研究精子细胞中的重组并分析突变率。

这一微流体设备分离了单个细胞并复制了它们的DNA
在单细胞研究的大潮中,新的测序方法层出不穷。不过,很少有人对这些方法进行系统的比对。慕尼黑大学生物学家Wolfgang Enard团队,在小鼠胚胎干细胞的基因表达研究中比较了一些常用的单细胞测序方法,包括Smart-seq、CEL-seq、SCRB-seq和Drop-seq。
01-Smart-seq
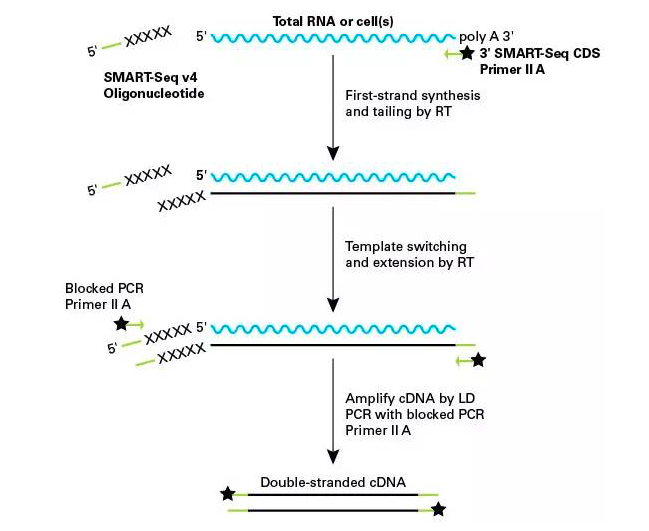
SMART(Switching mechanism at 5’ end of the RNA transc ript)是一个具有里程碑意义的重要技术。实际上,能够从单细胞生成全长cDNA的测序方案并不多,Smart-seq就是其中之一。对于等位基因特异性表达或者剪接变体研究来说,覆盖整个转录组是一件非常重要的事情。
Fluidigm C1单细胞制备系统能够自动完成Smart-seq步骤。你只需要将制备好的细胞悬液加进去,仪器就会分离并裂解细胞,把mRNA逆转录为cDNA,再对cDNA进行扩增。扩增后的cDNA可以拿来测序,也可以进行qPCR检测。
在Enard的比较研究中,Fluidigm C1系统的Smart-seq比较灵敏,成本也较高。这一系统使用的微流体芯片不能重复使用,不过这种芯片可以放到显微镜下观察,证实健康单细胞的存在。
02-CEL-seq
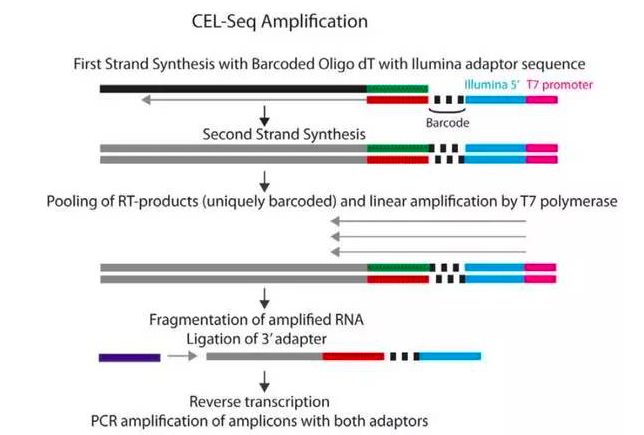
CEL-seq(Cell ex pression by linear amplification and sequencing)是一种采用线性扩增的常用测序方法。线性扩增的主要优势是错误率比较低,不过线性扩增和PCR都存在序列依赖性偏好。
CEL-seq技术发表于2012年,主要是分离单细胞,逆转录带有poly-A 尾巴的mRNA片段,给它们贴上代表其细胞来源的条码。2014年Science杂志发布的MARS-seq与CEL-seq很类似。
CEL-seq和其他线性扩增方法生成文库花费的时间要稍微长一点。不过,CEL-seq在早期阶段就给样本贴上条码并进行混合,大大减少了手动操作的时间。PCR是在实验的最后阶段使用的,主要是为了连接正确的测序接头。
CEL-seq所需试剂都是现成的,大约两天时间生成测序文库和测序数据,Yanai指出。需要注意的是,CEL-seq与大多数方案一样,测序转录本的3’端。Enard在实验研究中用到了CEL-seq数据,从这项研究来看,CEL-seq的重现性较好。
GitHub网站提供有CEL-seq可用的生物信息学工具。Yanai研究团队正在开发新版本CEL-seq2,新版本的灵敏度将比旧版本高三倍。
03-SCRB-seq
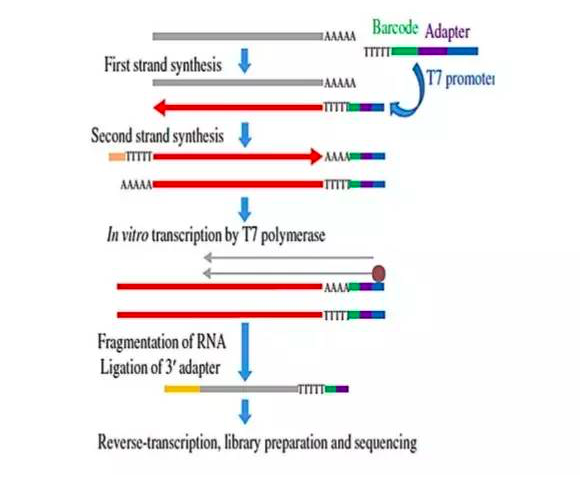
Broad研究所开发的SCRB-seq(single-cell RNA barcoding and sequencing)技术采用的是PCR扩增。该技术需要结合流式细胞仪(FACS)或者其他细胞分选方法,把单细胞分配到微孔里去。
SCRB-seq与Smart-seq比较相似,只不过SCRB-seq会整合特异性的细胞条码,以分辨扩增分子的来源,更准确的定量转录本。此外,SCRB-seq并不生成全长cDNA,而是像CEL-seq一样富集RNA 3’端。
2014年,他们将SCRB-seq技术提前发布在BioRXiv上。随后他们对这一技术进行了更新,并将其更名为“high-throughput eukaryote 3’ digital gene ex pression”。Broad研究所仍提供有SCRB-seq技术服务,SCRB-seq也被整合到了Wafer GenBio systems公司的scRNA-seq平台上。据说Fluidigm公司的C1系统很快也将支持SCRB-seq方案。SCRB-seq目前是Enard比较中意的测序方法,它不仅适用于单细胞RNA测序,还能支持大量细胞(bulk)的RNA测序。可惜的是SCRB-seq并不好DIY,研究者自己很难将测序流程与FACS对接起来。
04-DROP-seq和inDROP
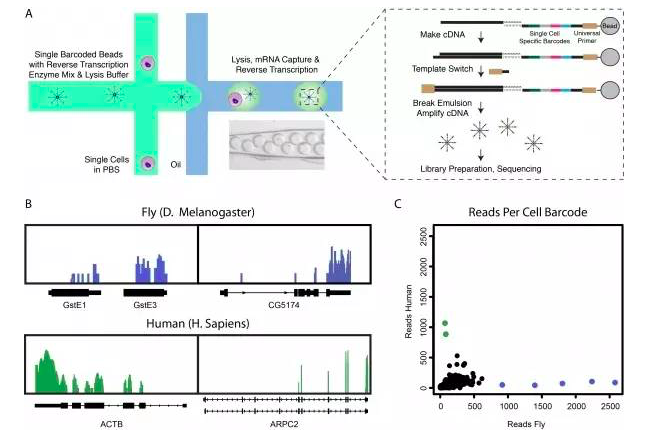
哈佛医学院的研究人员开发了以微滴为基础的两种独立技术Drop-seq和inDrop。他们利用微流体装置将带有条码的微珠和细胞一起装入微小的液滴,建立了快速、廉价、高通量的单细胞RNA-seq方法。这两种技术将细胞隔离在微小的液滴中,装上用于扩增的条码引物,由此检测数以千计的细胞。研究者们认为,Drop-seq和inDrop能够帮助生物学家进一步发现和分类人体细胞,绘制大脑等复杂组织的细胞多样性图谱,更好地了解干细胞分化,获得更多疾病的遗传学信息。
Enard及其同事发现,Drop-seq在单个细胞中检测的基因数还不到Smart-seq/C1、CEL-seq和SCRB-seq的一半。不过,在统计学水平上研究差异性表达的时候,高通量Drop-seq和SCRB-seq是比较划算的。哈佛医学院Steve McCarroll实验室去年下半年根据用户反馈,在自己网站上公布了3.1版的Drop-seq(mccarrolllab.com/dropseq)。
参与开发SCRB-seq的Stefan Semrau正在搭建自己的Drop-seq平台。Semrau从Whitehead生物医学研究所搬到Leiden物理研究所的时候,发现自己用FACS已经不那么方便了,于是他为新实验室选择了Drop-seq。建立微流体芯片和液滴系统是整个过程中比较艰难的部分,不过Semrau实验室的一名微流体博士后仅用三周就搞定了这些问题。Drop-seq文库制备是任何分子生物学研究者都熟悉的标准操作。据Semrau估计,搭建和优化Drop-seq大概只需要六个月时间。
05-scMT-seq
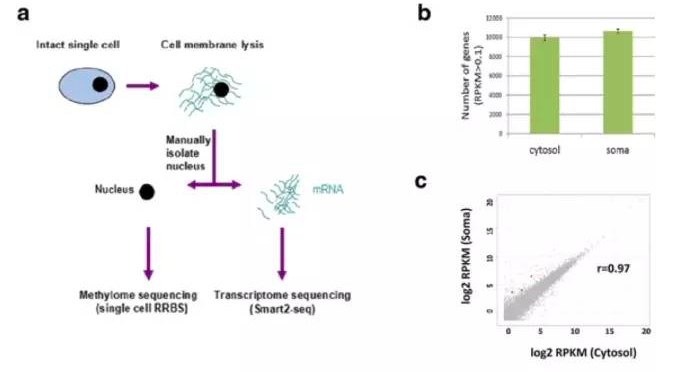
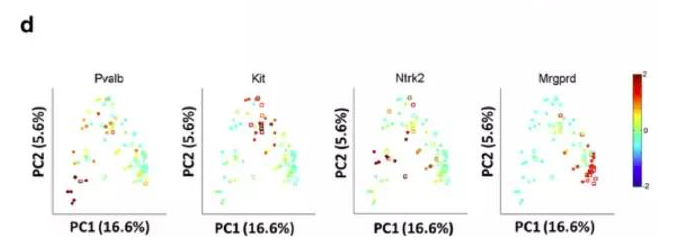
加州大学范国平教授和同济大学薛志刚教授在5月5日的Genome Biology杂志上发布了自己开发的新测序方法——scMT-seq。这种方法能够同时分析单个细胞的转录组和甲基化组。
研究人员用这种方法对感觉神经元进行研究,揭示了细胞间的转录组和甲基化组异质性。不过,这些差异大多不是启动子甲基化造成的。举例来说,启动子带CpG岛的基因,表达水平与基因体甲基化正相关。这项研究表明,scMT-seq可以用来检测单细胞中的转录组、甲基化组和单核苷酸多态性信息,进而解析表观遗传学基因调控机制。


