单细胞入门-读一篇scRNA-seq综述讲解:
1.1. 实验
简单回顾测序技术的发展,从桑格尔发明双脱氧末端终止法(一代测序)到人类基因组计划历时13年耗费30亿美元,测序一直很贵,直到高通量的边合成边测序技术(二代测序)出现。随着测序价格的不断下降,2009年开发出了单细胞转录组测序方法(汤富酬)。
经过8年多时间的发展,如今不同的scRNA-seq流程有了大量改进,它们一般都分为四步:
1. 单细胞(核)的分离和裂解
2. 反转录
3. cDNA扩增
4. 测序文库制备
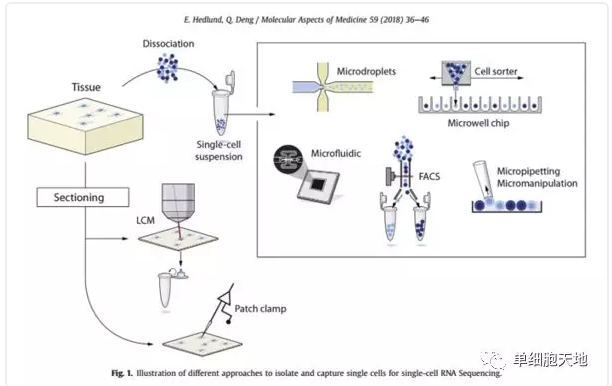
1.1.1. 单细胞分离的步骤至关重要
除游离细胞外的细胞分离,有两条路线:
i. 组织切片 - 激光捕获显微切割(LCM)或者 膜片钳(Patch clamp)
ii. 酶法去除细胞间质 - 各种微操技术分选出单个细胞(各有优劣)
微吸(Micro-pipetting)适用于细胞量少或比较珍贵的样品,通量低。
流式细胞分选(FACS)和微流控(Microfluidic)设备适用大量可用细胞,通量高。
● FACS同样用于筛选特定标记的某类细胞,它可能分出不止一个细胞和造成细胞损伤。
● 微流控更加温和,用于高度标准化的自动化流程,缺点是假定细胞损失和细胞大小偏好,目前的商用设备包括10X Genomics的Fluidigm C1系统和Illumina的Biorad SureCell系统(含ddSEQ细胞隔离器)。
微管平台(Microwell platforms)能够消除细胞大小偏好,也可以通过显微观察排除分出多个细胞的情况,商用设备有WaferGen的ICELL8单细胞系统。
多数单细胞收集方法都要求样品是完好的新鲜组织,因为微环境的改变影响正常细胞过程;酶促反应也可能使细胞产生应激,从而改变基因表达。有一个办法来避免这些问题,那就是只收集细胞核,细胞核包含未加工的mRNA和很少的mRNA。细胞核很黏,目前只有FACS能做到这一点。
1.1.2. 反转录
大部分公开的流程都是使用oligodT引物,可以捕获到具有多聚结构的mRNA和少部分lncRNA。
SUPeR-seq使用了混合oligodT和六碱基随机引物的方法,然而它没有去除rRNA却只检测到很少的rRNA,猜测是没有把二级结构打开。
MATQ-seq近期被报道比Smart-seq2更灵敏,产量更高。它是基于MALBAC引物设计的,能做到全基因覆盖,检测总RNA。
1.1.3. cDNA扩增
反转录结束后,有多种策略合成了第二条cDNA链
一种是SMART技术(switching mechanism at 5' end of RNA template)
这个系列包括Smart-seq,Smart-seq2,STRT,利用转移酶和小鼠白血病病毒反转录酶来进行链置换并加上后续PCR扩增的接头。
PCR是常用的指数扩增技术,很容易因为GC含量的差异造成扩增偏倚。
另一种就利用了体外转录的方式(IVT)进行线性扩增
这个系列包括CEL-seq,MARS-seq,CEL-seq2,通过将T7启动子连在oligodT引物上,可以在cDNA合成后启动IVT。IVT取消了对模板置换的需求。
另外,MALBAC-RNA使用准线性扩增,它的引物能生成末端互补的扩增子,形成闭环来防止指数复制。
1.1.4. 方法选择以及测多少细胞
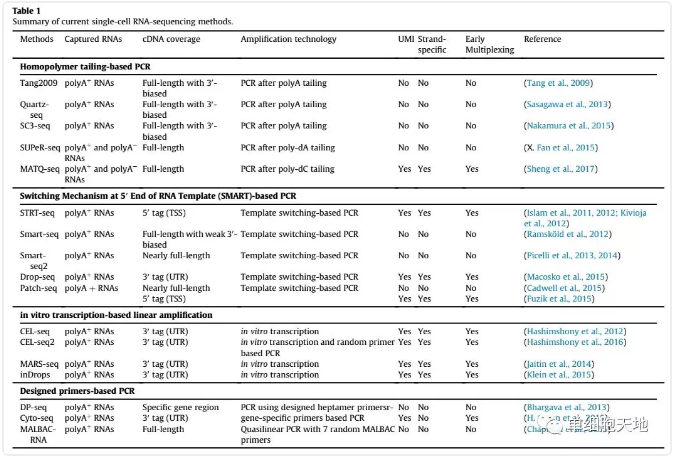
不同的技术流程按照cDNA覆盖大致可以分为两类:全长(full-length)和基于标签(tag-based)。
全长的方法试图得到基因体均匀读长覆盖并增加匹配序列数,更适合亚型发现、剪切事件、SNP鉴定等等分析。一大缺陷是建库通量较低,难以混样测序。更重要的是,它不能结合UMIs(unique molecule identifiers)来进行数字量化。有一个例外,MATQ-seq可以把barcodes和UMIs整合到MALBAC引物上,从而克服这个缺陷。
基于标签的方法可以继续细分成5'还是3',主要优点是能结合UMIs,可以混合多个样品,允许基因水平的定量优化。因为读长被限制在序列一端,相对而言灵敏度较低,大部分仅用于基因表达定量。
选择什么方法取决于要回答的生物学问题。如果是发现细胞类型和鉴别组织成分,两种方法都可以。基于标签的方法可以在反转录之后把所有样品混在一起,价格更便宜规模可以更大。如果是等位基因表达、不同亚型的发现,全长的方法更加合适。这些方法中,Smart-seq2在灵敏度和产量上都表现出众,不过要用到Tn5,比较贵,如果有很多很多的细胞要测,比如4000个,那么Drop-seq也是很好的选择。
关于灵敏度,需要考虑测序深度。这些方法都有一个共同点,当一个样品测到1M reads之后,灵敏度开始变得比较稳定,从1M reads 测到 4.5M reads,灵敏度只略微提升。
需要多少细胞的数据用来分析,取决于细胞类型的罕见程度。
Nicholas E. Navin提供了一个计算公式 P(d) =1-(1-s)^n
P(d):检出能力(detection power) s:等同于亚克隆频率(subclonal frequency) n:要测的细胞数
如果感兴趣的细胞亚型占比约为1%,需要测250个细胞使检出能力达到0.9,需要测500个细胞使检出能力达到1.0。另外也需要做重复实验来评估假阳性率和假阴性率。
需要的细胞数和必要的测序深度同样依赖于感兴趣的细胞与其他细胞的差异程度,如果这种细胞有非常独特的转录特征,那么测的细胞数少一点,测序深度浅一点也是可以的。
1.1.5. scRNA-seq的技术挑战
SingleCell的问题:细胞与细胞之间有很强的异质性。
只有一个细胞,初始数据量就小,噪音就大。
RNA捕获效率不稳定,文库制备的随机丢失会制造技术噪音。
随机基因表达,不同的细胞状态细胞大小细胞周期会产生生物噪音。
批次效应使高通量的实验数据存在系统误差。
认真规划实验步骤,作多次生物学重复可以降低批次效应,然而生物样品的遗传背景是很难通过实验步骤来控制的。
鉴定批次效应的一个办法是通过主成分分析(PCA),看细胞是否会按照相应的起源进行分群。
为了解释技术操作带来的误差,通常加入外源的RNA进行质控。不同浓度、长度、GC含量的合成RNA可以起到监控作用。
但是外源样品与内源RNA的分子特征并不会完全相同,对照作用有限。
怎么减少RNA损失,使信息能够保真是scRNA-seq的关键性挑战,测序结果仍需要谨慎对待,建议做功能性验证。
1.2. 应用
过去几年,scRNA-seq已被应用于发现新的细胞类型,探索动态发育过程,鉴定基因调控机制,揭示随机等位基因表达。
这篇综述只着重介绍了胚胎植入前发育和大脑皮层,在这两个方向上scRNA-seq有了巨大的概念性发展。
1.2.1. 胚胎植入前发育
生命起源于一个受精卵,受精卵的分化过程受转录水平调控形成三个主要的细胞谱系。这个过程里有几个长期存在的问题:1. 单个卵裂球之间是何时出现差异的?2. 三个细胞谱系如何及时分离?3. 胚胎基因组是何时激活的?4. 早期的规范化事件是否存在物种间差异?
scRNA-seq为这些问题的解答提供了新的思路。早先对小鼠胚胎的早期卵裂球进行实验操作(包括增、减单个细胞),都不会影响到胚胎发育,表明早期卵裂球会经历一个调节发育(受到感应信号可以变成任何细胞类型)。然而scRNA-seq的结果显示,早在四分体时期,卵裂球间已经存在分子不对称了。后来通过比较滋养外胚层(TE)和内细胞团(ICM)的细胞命运,鉴定出Sox21基因在四分体时期存在稳定的异质表达,并且影响后代细胞的分化路线。在植入前发育的各个阶段,通过scRNA-seq可以得到一个全过程的基因动态表达视图,跨物种数据比较发现人和小鼠的胚胎发育存在很多的生物学差异,如胚胎基因激活时间,细胞谱系建立时期,等位基因特异性表达情况等等。对人类胚胎细胞进行具体的功能研究比较困难,后面换成了相近的猕猴细胞。
1.2.2. 小鼠大脑皮层
在神经系统科学领域,对所有哺乳动物的神经细胞进行系统性分类是一个长期的目标。理解大脑的细胞构成有助于破译它的功能和连接性。不同的研究表明,对来自小鼠大脑不同区域的细胞做scRNA-seq,进行细胞分群,发现中间神经元具有更大的异质性,暗示中间神经元细胞具备更加复杂多样的功能。通过基因表达谱得到的细胞类型分类是否显著关联不同的功能性质还有待进一步的研究,这些实验的方法都显示有一定的偏好性。
为了让基因表达直接关联解剖、形态、功能的属性,两个实验室同时开发出了Patch-seq,这个技术把全细胞电生理膜片钳记录与scRNA-seq相结合。
其中一个实验室结合膜片钳和Smart-seq2,在新皮质L1外层分析了58个皮层细胞,这项研究使用了机器学习以不同的放电模式来进行细胞形态分类,结果跟来自基因表达谱的分群结果对应的很好。58个细胞分出两种细胞亚型,eNGCs和SBCs,重要的是,发现SBCs富集了四个神经精神病相关的基因。
另一项研究使用膜片钳和STRT-seq,在躯体感觉皮质的1/2层分析了45个中间神经元和38个椎体神经元细胞,根据电生理性质和形态,分为5个亚型和3个亚型。这八个亚型跟scRNA-seq鉴定到的分群结果相吻合,从而确认了Patch-seq方法的有效性。
Patch-seq的分析适用于离子通道和受体基因研究,可以预测神经生理学表型。跟鲜活细胞的scRNA-seq相比,Patch-seq捕获到的基因显然更少,通量相对更低,然而正因为有不同的单细胞测序方法,使得从单细胞尺度上深入分析分子特征、形态和异常复杂组织的功能成为可能。
1.3. 未来展望
1.3.1. 空间转录组
单分子原位荧光杂交技术(smFISH)在2008年被开发出来,用作单细胞尺度的组织RNA定量,它使用带荧光基团的20bp核酸探针。这项技术受限于能够同时检测到的转录本数量,后来引入分组探针文库的组合标签克服了这一缺陷。随着七种光转换染料和空间条码结合超分辨显微技术的使用,能够同时检测到的基因数进一步增加。高分辨率的显微镜能够识别结合了同一种探针实际序列不同的mRNA。接着,通过使用顺序轮的杂交、成像、探针剥离来给mRNA加条码,继续优化了该方法。smFISH的一大优势是杂交效率很高,能够检测到95%的mRNA。smFISH适用于剪切变异、染色体位点以及SNP。类似的,荧光原位RNA测序(FISSEQ)也使用基因特异的探针来读取空间基因表达。跟smFISH明显不同的是,FISSEQ的reads比RNA-seq还少很多,丰度不够。总体上看,以上这些原位荧光的方法想要覆盖整个转录组,都比较费时费力。
使用LCM的单细胞空间转录组方法已经被开发出来了。LCM可以从速冻组织切片中仔细分离出单个细胞,分辨率能达到亚细胞水平。LCM适用于任何胚胎和成熟时期,特别是那些难以分离的组织。通过简单的组织染色或者快速的抗体染色可以鉴定出感兴趣的细胞。LCM是与全转录组基因芯片结合,然后是RNA-seq,直到现在,需要的细胞数也是数百上千。结合scRNA-seq和LCM的LCM-seq,通过直接裂解分离的细胞,消除通常是在LCM之后的RNA隔离步骤,可以简化流程,降低技术噪音,减少费用。同时每个细胞的空间信息都保留了下来并且不需要组织分离步骤,从而能够在单细胞水平同时研究细胞异质性和空间差异。保留空间信息的重要性不应该被低估它可能是组织内细胞识别的关键性因素。此外,因为细胞在分离前保留了原有位置的连接信息,比起需要进行组织裂解的测序方法,更能够反映生物体内的真实情况。LCM-seq另一个优势是可以用于缺损和部分退化的组织。然而,至今为止的一大缺陷是RNA有一些片段化,即使处理的时间很短也一样,所以覆盖度比起鲜活细胞要低,不能作RNA剪切的深入分析。LCM染色的后续优化有可能克服这一障碍。
一种叫作”空间转录组(spatial transcriptomics)“的优雅方法近期被开发出来,能够不分离细胞直接使用完整的组织切片进行转录组分析。组织切片被放置在slide上,使用含有独特空间条码标记的反转录引物。 slide上布满直径100微米间隔200微米的孔,孔内有接近两亿个寡核苷酸探针。组织经过通透性处理后加上反转录试剂,组织被酶解,留下cDNA与slide上排列的探针结合。这种方法的分辨率很高,100微米,对于整体空间信息的接收在时间上非常高效。但是不容易显示出细胞的异质性,因为细胞大小的差异,这种方法只能展示出特定二维坐标下单一或多个图层的空间信息。

1.3.2. 单细胞多组学
测序技术目前已经能够从同一个细胞中获取基因组、表观组、转录组和蛋白组的情况。因此,可以整合每个细胞的DNA、RNA、蛋白还有表观修饰的信息得到一个综合的理解。为了这个目的开发的方法有:DR-seq和G&T-seq,同时分析基因组和转录组;scTrio-seq,基因组、转录组和甲基化谱;scM&T-seq,转录组和甲基化谱;PEA-qPCR,蛋白和一个基因panel。同时研究基因组和转录组可以在基因表达水平关联CNV、染色体混合和调控因子的SNV。还可以揭示克隆结构和细胞亚型,直接联系基因型和表型。另一方面,结合转录组和甲基化分析,可以知道单细胞中基因组不同功能因子的DNA甲基化水平与基因表达水平的关系。未来把总RNA,小RNA,染色体重组和结构结合到单细胞多组学里,可以更加详细的描述正常细胞功能和疾病过程。
另一个新兴的前沿研究是结合系统基因功能分析和scRNA-seq分析。
1.3.3. 人类细胞图谱和精准医学
2016年一群科学家开启了人类细胞图谱计划(Human Cell Atlas),目前已经包括了免疫系统、中枢神经系统、上皮组织、胚胎细胞和癌症。这个计划将会提供一个囊括了细胞类型、标记基因、信号通路和调控机制的综合参考视图,给不同个体和疾病的组织带来更好的生物靶标识别和药物标靶,从而进一步发展精准医学。
1.3.4. 把转录水平的差异关联到细胞类型和功能
scRNA-seq的数据已经表明,在大脑不同区域和不同的组织,细胞间的异质性比之前预计的还要大。面前更艰巨的任务是从功能上评估RNA成分的异质性具体在何种程度上影响了相关细胞表现出不同的功能。大部分的scRNA-seq研究对此有所描述,仍未清楚的是,多大程度的转录组差异会导致细胞功能的区别,使细胞成为不同的类型而不是同类型细胞的不同可选状态。某(几)种转录本的表达量积累到什么水平能够看到明显的细胞功能改变?这取决于该基因的功能以及其他的基因表达,还取决于特定转录本的稳定性和半衰期。不经过功能测试就将功能与转录水平关联起来不是一个简单的任务。无论如何,细胞和分子生物、生物化学、生理学以及数学模型的结合,将来肯定能够解答革命性的scRNA-seq技术还不能回答的问题。单细胞技术在未来的生物功能注释中将会是不可或缺的工具。
1.4. 分析
这篇综述没有提到具体的数据分析。
(文章来源: 单细胞天地 转载仅供参考学习及传递有用信息,版权归原作者所有,如侵犯权益,请联系删除)


